First author: Jian Zhang; Corresponding author: Jing He, DOI:10.1002/anie.202312068
Carbonylation reactions, introducing one or two carbonyl groups into the parent compounds, have already been recognized as one of powerful protocols for constructing carbonyl-containing compounds that have wide application in industry to create pharmaceuticals, agrochemicals and their intermediates. Carbonylation of ethanol provides a sustainable and economical route to build value-added ethyl formate, diethyl carbonate, and diethyl oxalate. Among of these esters, ethyl formate has been not only applied widely in synthetic chemistry but also as FDA-approved food-flavoring fumigant.
In carbonylation reactions, CO gas has extensively been employed as the carbonyl source owing to its relatively low price, abundance, and versatility. But CO2 is more attractive as carbonyl source owing to its nontoxicity and nonflammability compared to CO. However, the thermodynamic stability and kinetic inertia of CO2 makes the activation of CO2 of great challenge. Especially, the endothermic CO2 to CO process (ΔH298 K = 42.1 kJ mol-1) requires a high working temperature to achieve a satisfied CO2 conversion (~ 50% of CO2 equilibrium conversion normally requires at least 600 oC). Such high temperature could easily cause the cleavageof -C-H, C-O, and even C-C bond to convert ethanol to acetaldehyde, CO2, or even coke deposition, irreversibly deteriorating the specific activation of alcoholic O-H bond, the absolute prerequisite for ethanol carbonylation to ester C-O bonds. To accomplish the carbonylation using CO2 as carbonyl source, CO2 reduction to CO and then CO carbonylation are often necessary.
Herein, Prof. Jing He proposed a photo-thermal cooperative strategy for carbonylation of ethanol with CO2, in which thermally-catalyzed dissociation of alcoholic O-H bond by the oxygen vacancies (Ov) on SrTiCuO3-x provides not only the carbonylation counterpart to reactive *CO, but also provides active *H for the photo-driven activation of CO2 on Cu2O-SrTiCuO3-x interfacial sites to reactive *CO (Scheme 1). A formation rate of 320 μmol g-1 h-1 to EF with a selectivity of 85.6% to the targeted activation of alcoholic O-H bond has been achieved in photo-thermal assisted gas-solid process under 3.29 W cm-1 of UV-Vis light irradiation (144 oC) and 0.2 MPa CO2.

Scheme 1. Illustration of photo-thermal cooperation in carbonylation of ethanol with CO2 on Cu2O-SrTiCuO3-x interface under UV-Vis light irradiation.
In-situ DRIFTS and XPS of CO2 adsorption has revealed that TiIII and interfacial CuI-O-TiIII sites are responsible for CO2 adsorption and ethanoladsorption and ethanol/ CO2 competitive adsorption demonstrates the Ov provides active sites for ethanol adsorption. Under light irradiation and active *H formed by the dissociation of alcoholic O-H bonds, the cleavage of C-O bond in CO2 adsorbed at the interfacial CuI-O-TiIII sites occurs, generating active *CO. Then the active *CO couples with the *OC2H5 formed by the dissociation of ethanol O-H bonds, forming ethyl formate. The formation rate of ethyl formate increases with the increase of the interface site. Furthermore, isotope labeling experiments has been applied to confirm the direct carbonylation mechanism of CO2. This strategy can be further extended to other low-carbon alcohols carbonylation, such as methanol, affording not only methyl formate but also dimethyl carbonate. This work provides new insight for the conversion of CO2 to value-added chemicals.
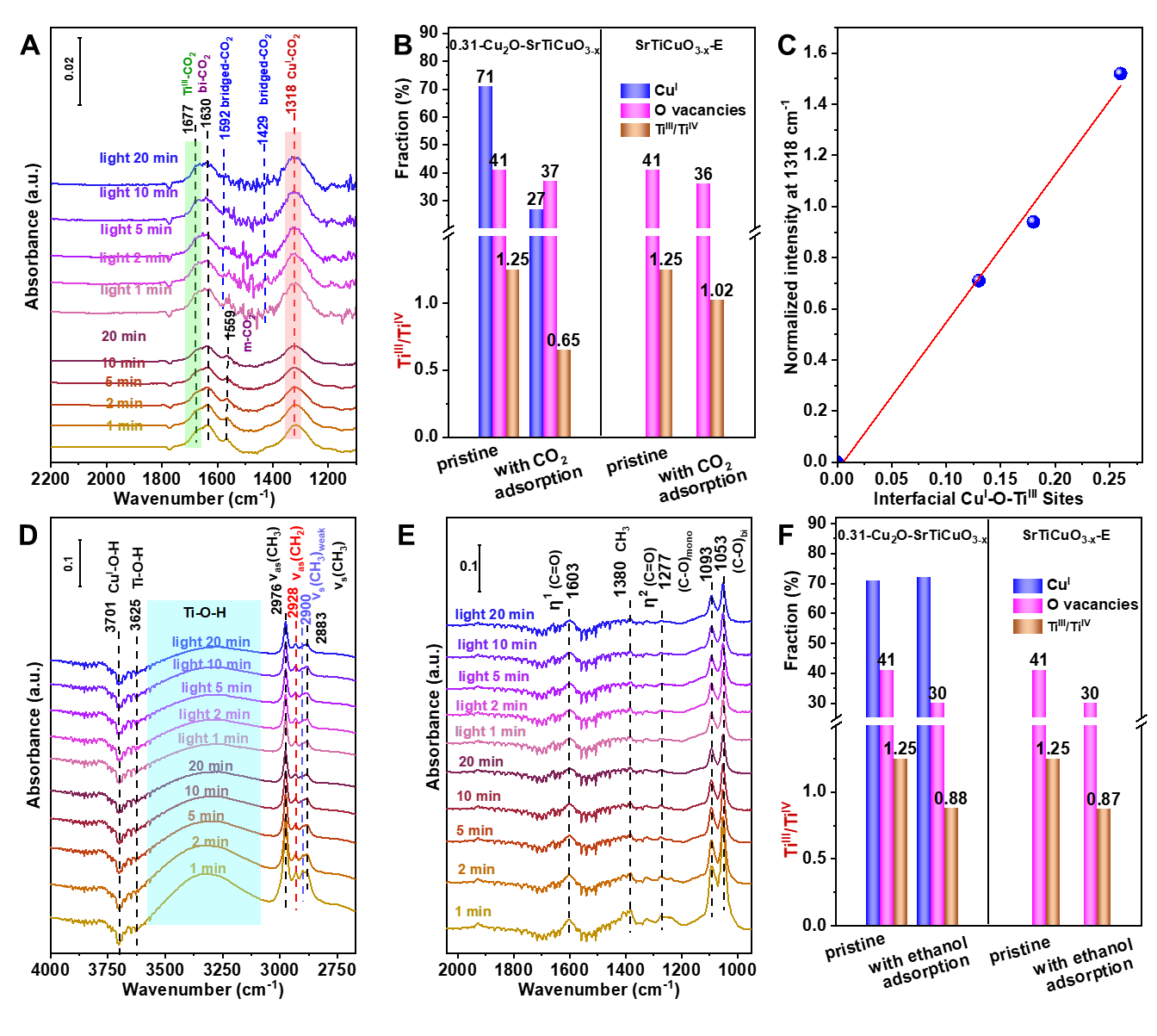
Figure 1. In-situ DRIFTS of (A-C) CO2 adsorption and (D-F) ethanol adsorption
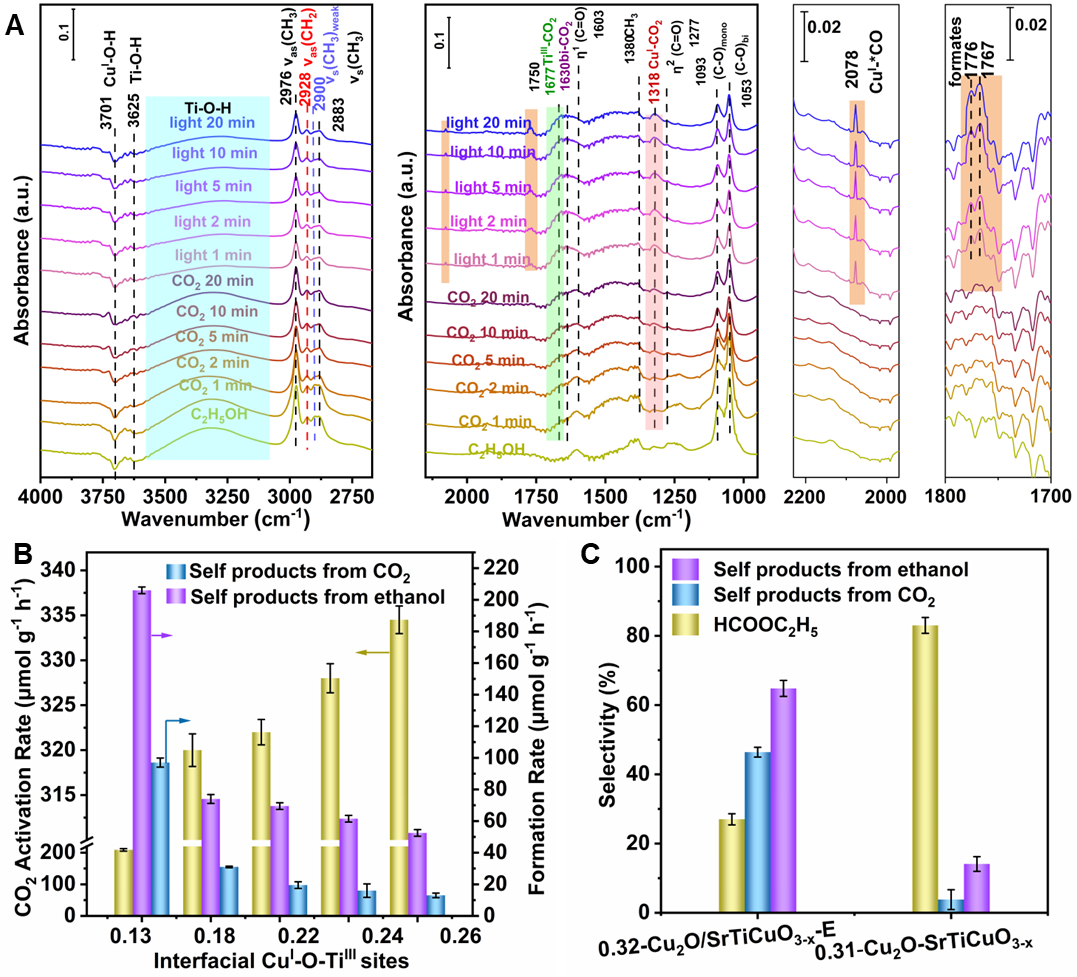
Figure 2. Ethanol/CO2 carbonylation
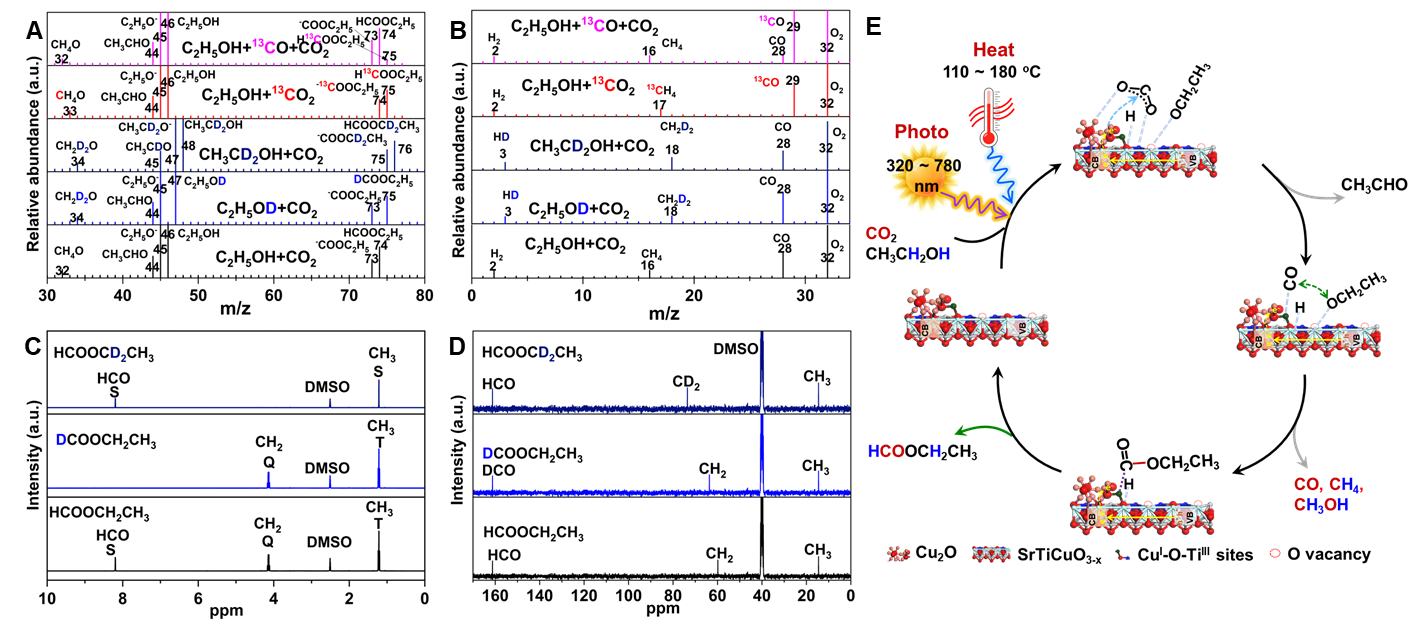
Figure 3. Ethanol/CO2 carbonylation mechanism